Welcome to msRepDB database

msRepDB - a comprehensive multi-species repetitive sequence database
Authors: Xingyu Liao, Kang Hu, Adil Salhi1, Jianxin Wang and Xin Gao
Corresponding Author: Prof. Xin Gao (xin.gao@kaust.edu.sa)
Repetitive sequences are prevalent in the genomes of all bacteria, plants and animals, and they cover nearly half of the human genome (01,02). Repetitive sequences play indispensable roles in the evolution, inheritance, variation, genomic instability, and serve as substrates for chromosomal rearrangements that include disease-causing deletions, inversions, and translocations (03,04,05,06,07). For example, the number and types of repetitive sequences vary between organisms and may reflect how rapidly an organism evolves to changes in its environment (08,09). Comprehensive identification, classification and annotation of repetitive elements in genomes can provide accurate and targeted solutions for research and diagnosis of complex diseases, optimization of plant properties, development of new drugs, and individual health management. RepBase (10) and Dfam (11) libraries are two most frequently used repeat databases, but they are not sufficiently complete. For instance, in the Drosophila genome, when the combination of RepBase and Dfam is used as the repetitive sequence database, only 4.04% of bases can be accurately annotated as repetitive sequences, as compared to 23.39% for msRepDB. However, it is well known that the proportion of repeat in Drosophila genome should be about 22%, which means that there are a large number of repeats cannot be annotated. Due to the lack of a comprehensive repetitive sequence database of multiple species, the current research in this field is far from being satisfactory.
LongRepMarker (12, DOI:10.1093/nar/gkab563) is a new framework developed recently by our group for comprehensive identification of genomic repetitive sequences. By integrating the detection results of LongRepMarker and existing databases (i.e, RepBase and Dfam), we here propose msRepDB, which is currently the most comprehensive multi-species repetitive sequence database (i.e., it contains 61,347 species). msRepDB takes the reference sequence or assembly of species as the input, and generates the masked sequence and comprehensive annotation report as the output. When the input data are reference sequences or assemblies, it should be in the fasta format (https://en.wikipedia.org/wiki/FASTA_format), and msRepDB takes out all the sequences and matches them with the database to find out the repeated elements contained in the sequences, as well as their locations and types, and finally masks the repeated elements in the input sequence and generates an annotation report. msRepDB also provides query and download functions. Users can retrieve and download the repetitive elements and their annotation reports from msRepDB according to the axon name or family name. On the other hand, if the user does not have any data, but just a taxon name or a repeat family name, msRepDB will also retrieve all relevant contents from the database and provide download links.
We deployed the first working version of msRepDB on the university intranet in October, 2020. Since then, msRepDB has handled more than 100 jobs to evaluate its performance and functionality, as well as its compatibility with major web browsers. We have had six individuals outside of our group with different background to test the database. We have conducted various experimental evaluations on the comprehensiveness of the msRepDB database. For example, we used the latest version of RepeatMasker (V.4.1.2) to classify and annotate the repeats of rice and drosophila based on msRepDB and the combination of the latest RepBase (V.26.06) and Dfam (V.3.3). The experimental results show that RepeatMasker annotated 315,702 DNA-type repeats (17,435.133kb in length) on the rice genome based on msRepDB, as compared to 8,008 DNA-type repeats (247.292kb in length) for the combination of the state-of-the-art databases. All the experimental data will be shown in the manuscript.
Main improvements of msRepDB

Compared with the existing repeat databases, the major improvements of msRepDB are as follows:
1. msRepDB contains more species than RepBase and Dfam databases
RepBase and Dfam libraries are the two most frequently used repeat databases, but they are not
sufficiently complete, because most of the repetitive sequences collected in these two libraries are
obtained through some existing detection methods (such as RepeatMasker, RepeatScout, RepeatModeler
and RepeatModeler2). Due to the limitations of sequencing data and the defects in design of the
detection principle, existing detection methods cannot accurately and comprehensively obtain the
repetitive sequences of species.
LongRepMarker (DOI:10.1093/nar/gkab563, https://github.com/BioinformaticsCSU/LongRepMarker)
is a new framework developed recently by our group for comprehensive identification of genomic
repetitive sequences. The comprehensive experiments carried out in the study of LongRepMarker not
only show that LongRepMarker can achieve more satisfactory results than the existing detection
methods (Table 1), but also can discover a large number of new repeat sequences and families
(Table 2). By integrating the detection results of LongRepMarker and existing databases (i.e,
RepBase and Dfam), we here propose msRepDB, which is currently the most comprehensive multi-species
repetitive sequence database (i.e., it contains more than 62,000 species).
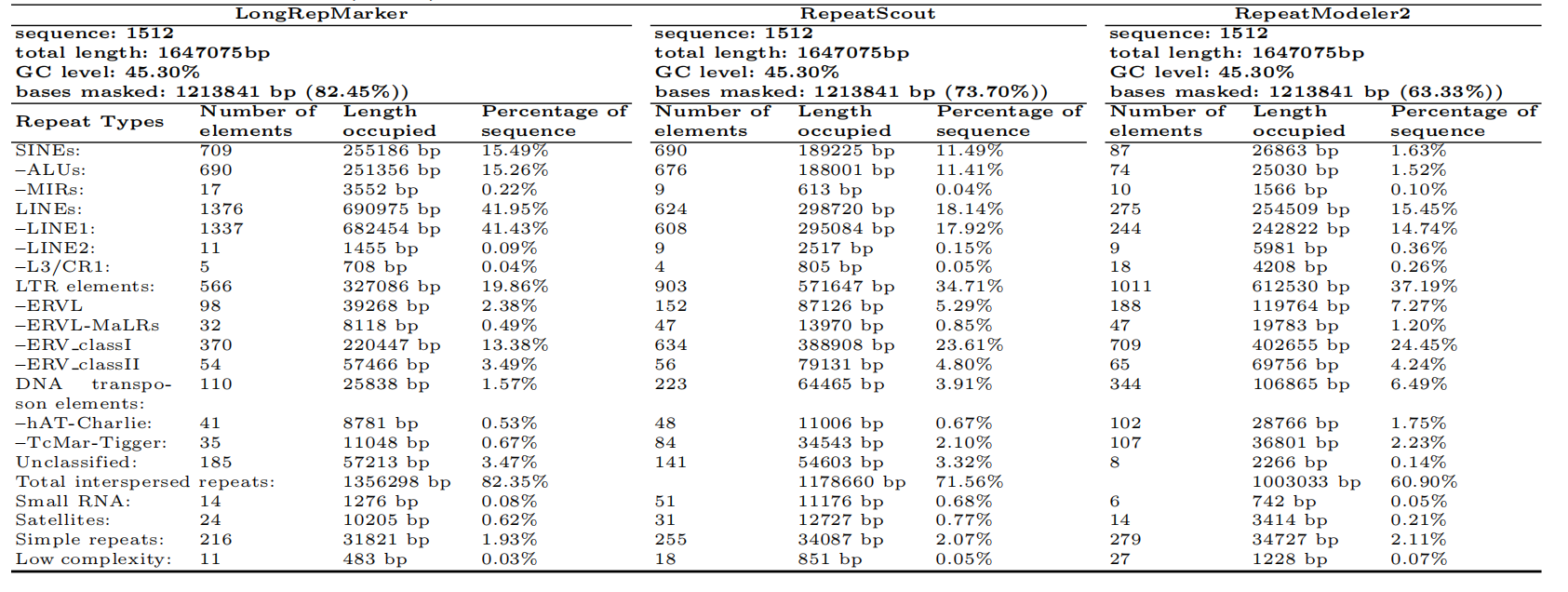
Table 1. Comparison of the proportion and detailed classification of detection results generated by three tools on Human(hg38) dataset covering the corresponding RepBase library.
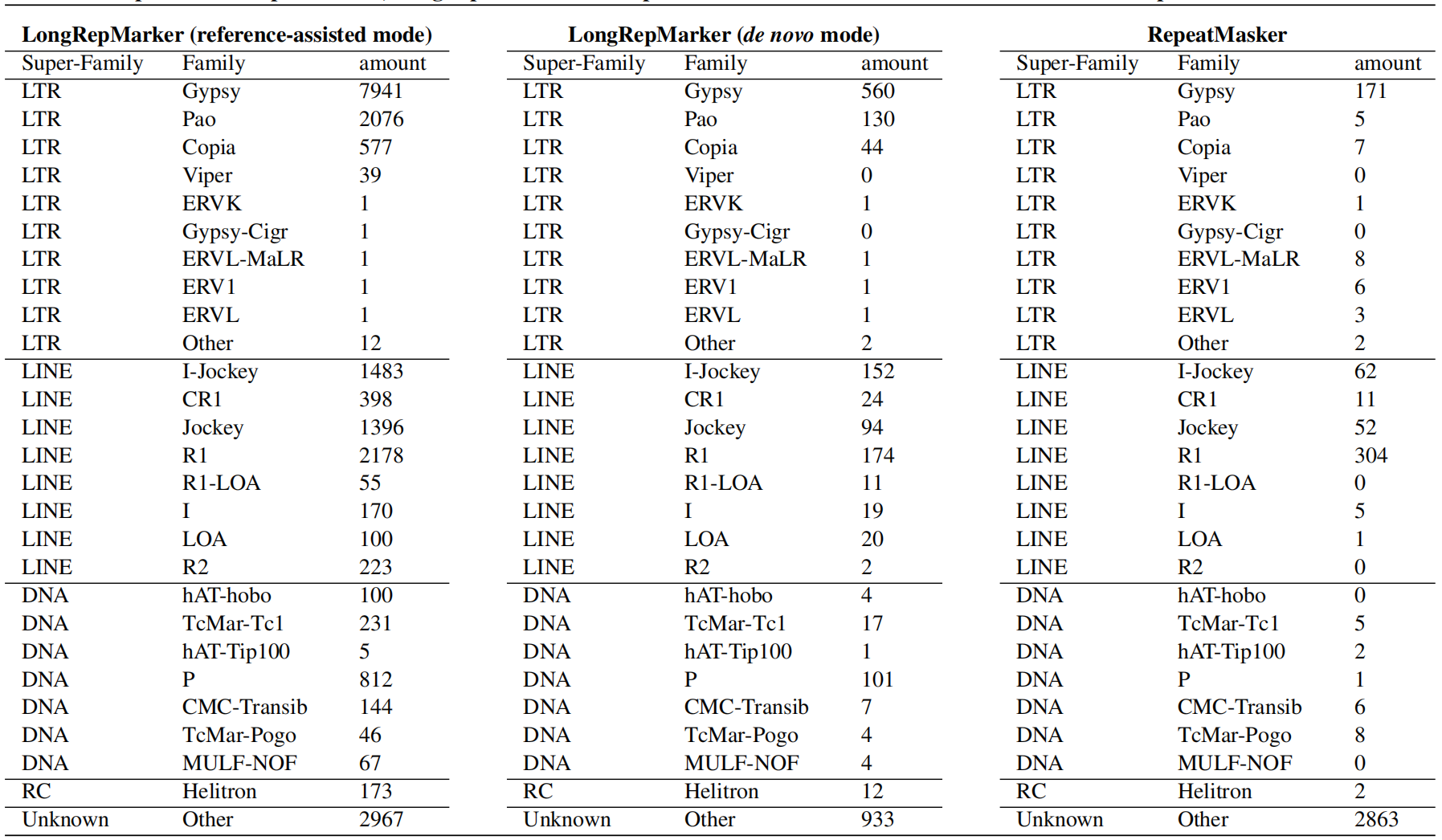
Table 2. Compared with RepeatMasker, LongRepMarker found repeat families and their detailed numbers on the Drosophila dataset.
2. For a single species, msRepDB contains more complete repeats and families than the existing repeat databases
We have conducted various experimental evaluations on the comprehensiveness of the msRepDB database. For example, we used the latest version of RepeatMasker (V.4.1.2) to classify and annotate the repeats of rice and drosophila based on msRepDB and the combination of the latest RepBase (V.26.06) and Dfam (V.3.3). The experimental results show that RepeatMasker annotated 80,003 LINE-type repeats (8,110.286kb in length) on the drosophila genome based on msRepDB, as compared to 49,524 LINE-type repeats (82.968kb in length) for the combination of the state-of-the-art databases (Table 3), and annotated 315,702 DNA-type repeats (17,435.133kb in length) on the rice genome based on msRepDB, as compared to 8,008 DNA-type repeats (247.292kb in length) for the combination of the state-of-the-art databases (Table 4). All the experimental data will be shown in the manuscript.
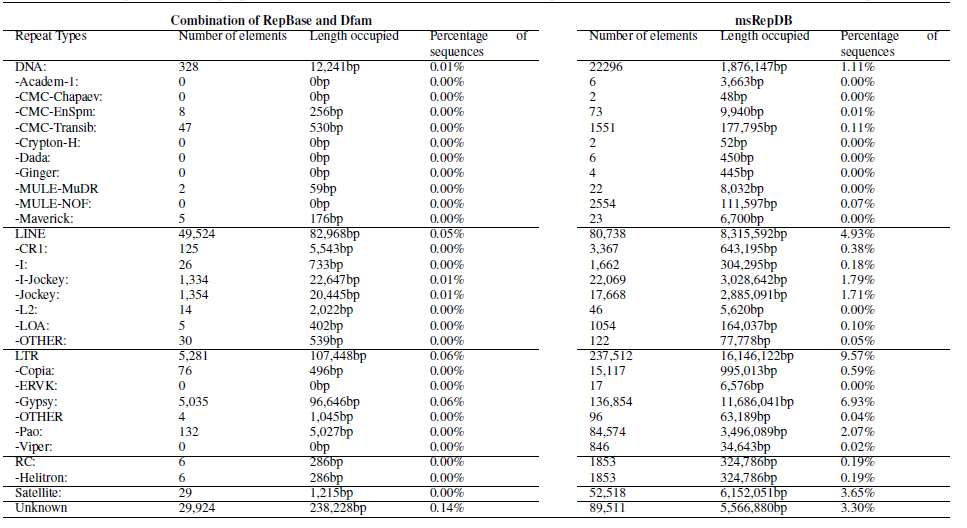
Table 3. Partial comparison of the proportion and detailed classification of detected repeats generated based on two databases of drosophila genome.
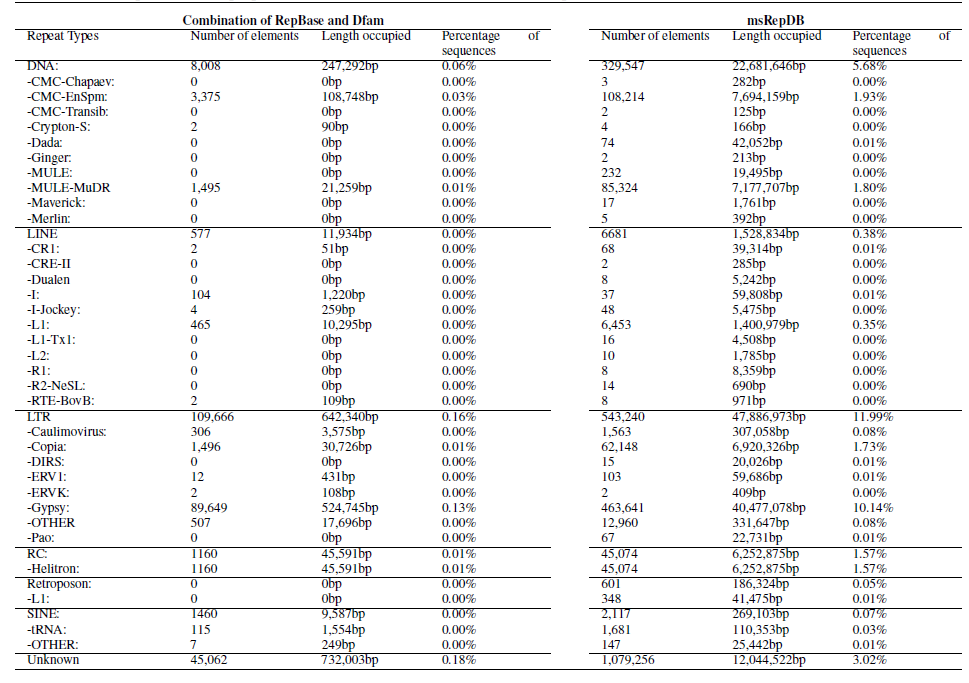
Table 4. Partial comparison of the proportion and detailed classification of detected repeats generated based on two databases of rice genome.
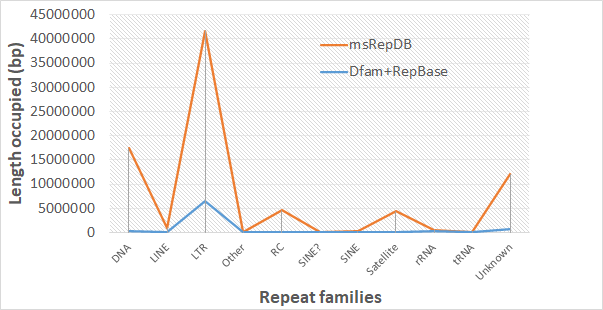
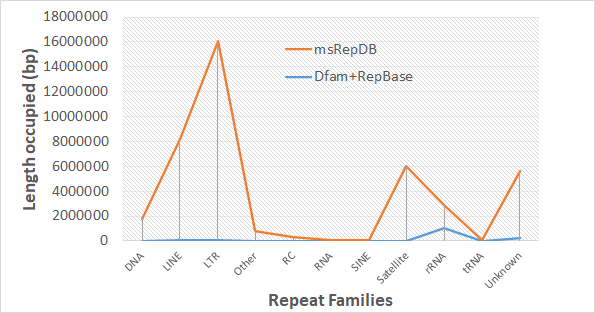
(A) Drosophila (B) Rice
Fig. 1 Comparison of the length occupied of repeat families based on two databases. Sub-figure(A) shows the comparison of the length occupied of repeat families of the two databases on the drosophila genome; Sub-figure(B) shows the comparison of the length occupied of repeat families of the two databases on the rice genome.
It can be seen from the experimental results shown above that msRepDB is the most complete multi-species repetitive sequence database at present because it integrates the detection results of longrepmarker, RepBase and Dfam libraries. We hope to be able to release the database for many species to the scientific community soon to benefit the genome research.
References
- Kazazian, H.H. Jr. (2004) Mobile elements: drivers of genome evolution. Science, 303, 1626-1632.
- Treangen,T.J. and Salzberg, S., L. (2011) Repetitive DNA and next-generation sequencing: computational challenges and solutions. Nature reviews. Genetics, 13, 36-46.
- Lu,Q., Wallrath,L.L.,Granok,H. and Elgin,S.C. (1993) (CT)n(GA)n repeats and heat shock elements have distinct roles in chromatin structure and transcriptional activation of the Drosophila hsp26 gene. Molecular and cellular biology, 13, 28022814.
- Kundu,T.K. and Rao,M.R. (1999) CpG islands in chromatin organization and gene expression. J. Biochem, 125, 217-222.
- Shapiro J.A. and von Sternberg R. (2005) Why repetitive DNA is essential to genome function. Biol. Rev., 80, 227-250.
- Kaltenegger E., Leng S. and Heyl A. (2018) The effects of repeated whole genome duplication events on the evolution of cytokinin signaling pathway. BMC Evol. Biol., 18, 76-95.
- Aguilera,A. and Garca-Muse T. (2013) Causes of genome instability. Annu. Rev. Genet., 47,1-32.
- George C.M. and Alani E. (2012) Multiple cellular mechanisms prevent chromosomal rearrangements involving repetitive DNA. Critical reviews in biochemistry and molecular biology, 47, 297-313.
- Hall A.C., Ostrowski L.A., Pietrobon V. and Mekhail,K. (2017) Repetitive DNA loci and their modulation by the non-canonical nucleic acid structures R-loops and G-quadruplexes. Nucleus, 8, 162181.
- Bao W., Kojima K.K. and Kohany O. (2015) Repbase Update, a database of repetitive elements in eukaryotic genomes. Mobile DNA, 6, 11-17.
- Storer, J., Hubley, R., Rosen, J. et al. (2021) The Dfam community resource of transposable element families, sequence models, and genome annotations. Mobile DNA, 12, 2-16.
- Xingyu Liao, Min Li, Kang Hu, Fang-Xiang Wu, Xin Gao and Jianxin Wang. (2021) A sensitive repeat identification framework based on short and long reads. Nucleic Acids Research, gkab563.
- Cox R. and S. M. Mirkin. (1997) Characteristic enrichment of DNA repeats in different genomes. Proceedings of the National Academy of Sciences of the United States of America, 94, 5237-5242.
- J. Yuyang Lu, Wen Shao, Lei Chang, Yafei Yin, Tong Li, Hui Zhang, Yantao Hong, Michelle Percharde, Lerui Guo, Zhongyang Wu et al. (2020) Genomic Repeats Categorize Genes with Distinct Functions for Orchestrated Regulation. Cell Reports, 30, 3296-3311.e5.
- Ahmad S. F., Singchat W., Jehangir M., Suntronpong A., Panthum T., Malaivijitnond S. and Srikulnath K. (2020) Dark Matter of Primate Genomes: Satellite DNA Repeats and Their Evolutionary Dynamics. Cells, 9, 2714.
- Lu S., Wang G., Bacolla A., Zhao J., Spitser S. and Vasquez K.M. (2015) Short Inverted Repeats Are Hotspots for Genetic Instability: Relevance to Cancer Genomes. Cell Reports, 10, 1674-1680.
- Shweta Mehrotra and Vinod Goyal. (2014) Repetitive Sequences in Plant Nuclear DNA: Types, Distribution, Evolution and Function. Genomics, Proteomics & Bioinformatics, 12, 164-171.
- Hannan A. (2018) Tandem repeats mediating genetic plasticity in health and disease. Nature Reviews Genetics, 19, 286-298.
- Mariely DeJesus-Hernandez, Ian R. Mackenzie, Bradley F. Boeve, Adam L. Boxer, Matt Baker, Nicola J. Rutherford, Alexandra M. Nicholson, NiCole A. Finch, Heather Flynn, Jennifer Adamson et al. (2011) Expanded GGGGCC hexanucleotide repeat in noncoding region of C9ORF72 causes chromosome 9p-Linked FTD and ALS. Neuron, 72, 245-256.
- Alan E. Renton, Elisa Majounie, Adrian Waite, Javier Simn-Snchez, Sara Rollinson, J. Raphael Gibbs, Jennifer C. Schymick, Hannu Laaksovirta, John C. van Swieten, Liisa Myllykangas et al. (2011) A hexanucleotide repeat expansion in C9ORF72 is the cause of chromosome 9p21-linked ALS-FTD. Neuron, 72, 257-258.
- Trost B., Engchuan W., Nguyen C.M., Thiruvahindrapuram B., Dolzhenko E., Backstrom l., Mirceta M., Mojarad B. A., Y. Yin, Dov A. et al. (2020) Genome-wide detection of tandem DNA repeats that are expanded in autism. Nature, 586, 8086.
- Mitra I., Huang B., Mousavi N., Ma M., Lamkin M., Yanicky R., Shleizer-Burko S., Lohmueller K. E. and Gymrek M. et al. (2021) Patterns of de novo tandem repeat mutations and their role in autism. Nature, 589, 246250.
- Hannan A.J. (2021) Repeat DNA expands our understanding of autism spectrum disorder. Nature, 589, 200-202.
- Beck C.R., Garcia-Perez J.L., Badge R.M. and Moran J.V. (2011) LINE-1 elements in structural variation and disease. Annu Rev Genomics Hum Genet, 12, 187-215.
- Chnais B. (2013) Transposable elements and human cancer: a causal relationship? Biochim. Biophys. Acta, 1835, 28-35.
- Belancio V. P., Roy-Engel A. M. and Deininger P. L. (2010) All y'all need to know 'bout retroelements in cancer. Semin. Cancer Biol., 20, 200-210.
- Hubley R., Finn R. D., Clements J., Eddy S. R., Jones T. A., Bao W., Smit A. F., and Wheeler T. J. (2016) The Dfam database of repetitive DNA families. Nucleic acids research, 44, D81D89.
- Price A.L., Jones N.C., Pevzner P.A. (2005) De novo identification of repeat families in large genomes. Bioinformatics, 21, i351-i358. Smit A.F.A., Hubley,R. and Green,P. RepeatMasker Open-4.0, 1996-2015.
- Schmutz J., Cannon S., Schlueter J., Ma J., Mitros T., Nelson W., Hyten D.L., Song Q., Thelen J.J., Cheng J. et al. (2010) Genome sequence of the palaeopolyploid soybean. Nature, 463, 178183.
- Jullien M. Flynn, Robert Hubley, Clment Goubert, Jeb Rosen, Andrew G. Clark, Cdric Feschotte and Arian F. Smit. (2020) Repeat- Modeler2 for automated genomic discovery of transposable element families. Proceedings of the National Academy of Sciences, 117, 9451-9457.
- Xingyu Liao, Min Li, Junwei Luo, You Zou, Fang-Xiang Wu, Yi Pan, Feng Luo and Jianxin Wang. (2020) Improving de novo Assembly Based on Read Classification. IEEE/ACM Transactions on Computational Biology and Bioinformatics, 17, 177-188.
- Xingyu Liao, Min Li, You Zou, Fang-Xiang Wu, Yi Pan and Jianxin Wang. (2020) An Efficient Trimming Algorithm based on Multi- Feature Fusion Scoring Model for NGS Data.IEEE/ACM Transactions on Computational Biology and Bioinformatics, 17, 728-738.
- Clausen P.T.L.C., Aarestrup F.M. and Lund O. (2018) Rapid and precise alignment of raw reads against redundant databases with KMA. BMC Bioinformatics, 19, 307.
- Koch P., Platzer M., and Downie B. R. (2014) RepARK{de novo creation of repeat libraries from whole-genome NGS reads. Nucleic acids research, 42, e80.
- Chu Chong, Rasmus Nielsen and Yufeng Wu. (2016) REPdenovo: inferring de novo repeat motifs from short sequence reads. PloS one, 11, e0150719.
- Xingyu Liao, Xin Gao, Xiankai Zhang, Fang-Xiang Wu and Jianxin Wang. (2020) RepAHR: an improved approach for de novo repeat identification by assembly of the high-frequency reads. BMC Bioinformatics, 21, 463.
- Xingyu Liao, Min Li, You Zou, Fang-Xiang Wu, Yi Pan and Jianxin Wang. (2019) Current challenges and solutions of de novo assembly. Quantitative Biology, 7, 90109.
- Sohn J. I. and Nam J. W. (2018) The present and future of de novo whole-genome assembly. Brief. Bioinformatics, 19, 2340.
- Qingyu Chen, Justin Zobel, Karin Verspoor. (2017) Duplicates, redundancies and inconsistencies in the primary nucleotide databases: a descriptive study. Database, 2017, baw163.
- Page A.J., Taylor B., Delaney A.J., Soares J., Seemann T., Keane J.A. and Harris S.R. (2016) SNP-sites: rapid efficient extraction of SNPs from multi-FASTA alignments. Microb Genom, 2, e000056.
- Bao Z. and Eddy SR. (2002) Automated de novo identification of repeat sequence families in sequenced genomes. Genome Res, 12, 1269-1276.
- Li H. and Durbin R. (2009) Fast and accurate short read alignment with Burrows-Wheeler Transform. Bioinformatics, 25, 1754-60.
- Li Heng. (2018) Minimap2: pairwise alignment for nucleotide sequences. Bioinformatics, 34, 3094-3100.
- Hannan A.J. (2010) Tandem repeat polymorphisms: modulators of disease susceptibility and candidates for 'missing heritability'. Trends Genet, 26, 59-65.
- C. M. Everett and N. W. Wood. (2004) Trinucleotide repeats and neurodegenerative disease. Brain, 127, 23852405.
- Lynch-Sutherland C.F., Chatterjee A., Stockwell P.A., Eccles M.R., Macaulay E.C. (2020) Reawakening the Developmental Origins of Cancer Through Transposable Elements. Front Oncol, 10, 468483.
- Chenais B. (2015) Transposable elements in cancer and other human diseases. Curr Cancer Drug Targets, 15, 227-242.
- Baillie J.K., Barnett M.W., Upton K.R., Gerhardt D.J, Richmond T.A., Sapio F.D., Brennan P.M., Rizzu P., Smith S., Fell M., Talbot R.T., Gustincich S. (2011) Somatic retrotransposition alters the genetic landscape of the human brain. Nature, 479, 534-537.
- Zhang X., Zhang R. and Yu J. (2020) New Understanding of the Relevant Role of LINE-1 Retrotransposition in Human Disease and Immune Modulation. Front. Cell Dev. Biol., 8, 657.
- Solyom S., Ewing A.D., Rahrmann E.P., Doucet T., Nelson H.H, Burns M.B, Harris R.S., Sigmon D.F., Casella A., Erlanger B. et al., (2012) Extensive somatic L1 retrotransposition in colorectal tumors. Genome Res, 22, 2328-2338.
- Scott E.C., Gardner E.J., Masood A., Chuang N.T., Vertino P.M., Devine S.E. (2016) A hot L1 retrotransposon evades somatic repression and initiates human colorectal cancer. Genome Res, 26, 745-755.